The Max Planck Institute Reconstructs Key Protein Complexes
"Parallel Computing Toolbox enabled us to speed up our processing by 20 to 30 times. We were able to use our cluster productively from the MATLAB environment without having to be experts in parallel programming or having to learn another programming language."
Challenge
Solution
Results
- Research time cut by years
- Development time cut from weeks to days
- Workflow accelerated
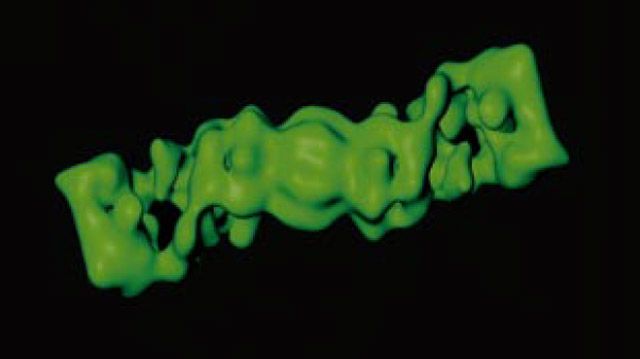
Protein degradation is a key mechanism for controlling a variety of cellular functions and pathways. One critical pathway, protein breakdown, is regulated by the 26S proteasome. As part of a cell’s central mechanism for protein degradation, the 26S proteasome could become a key molecular compound for cancer therapies. However, the instability of the 26S complex and its consequent dissociation into minor subcomplexes make it a difficult structure to elucidate.
Structural and computational biologists at the Max Planck Institute of Biochemistry have reconstructed the 26S and other key protein complexes in 3D from 2D projections obtained by cryo-electron microscopy. This work was made possible by the streamlined procedures for image acquisition, filtering, processing, and 3D reconstruction that the researchers developed using MathWorks tools.
"With MathWorks tools we can conduct the entire workflow in a single environment," says Andreas Korinek, scientist at the Max Planck Institute of Biochemistry. "Instead of converting coordinates and data between five or six different packages, we can use one platform for controlling instruments, acquiring and filtering images, and constructing 3D structures in an accelerated process."
Challenge
In cryo-electron microscopy, researchers capture 2D projections of ice-embedded protein samples kept at liquid nitrogen temperatures. The individual protein complexes assume random orientations within the ice, providing the angular sampling required to reconstruct a 3D structure from all angles. To minimize beam damage to these sensitive samples, researchers apply relatively low electron doses during imaging.
To calculate a reconstruction of the protein structure, researchers must produce class averages corresponding to individual projections of the 3D structure with high contrast and signal-to-noise ratios. Because the electron doses reduce the contrast and signal strength of the imaged projections, potentially millions of projections must be averaged to achieve the required image quality.
Given the large numbers of individual projections required, scientists at the Max Planck Institute needed to develop high-throughput tools and procedures capable of accurately processing vast amounts of data.
Solution
Max Planck Institute researchers used MathWorks tools to control the electron microscope, automatically select individual projections from micrographs, average and process projections, and reconstruct an accurate 3D density map of the protein complex.
They used a graphical user interface (GUI) developed with MATLAB® to automatically collect the large numbers of individual projections required.
The electron microscope first collected low-magnification survey images of the entire sample grid. The researchers used these images to identify areas of sufficiently thin ice-containing samples. A fully automated acquisition procedure then collected high-magnification images from these samples.
In electron microscopy, numerous imaging factors (collectively referred to as the contrast transfer function) contribute to image formation. As a result, different frequencies are recorded at varying sensitivities. Max Planck used Image Processing Toolbox™ to correct for these perturbations to the data. Using Statistics and Machine Learning Toolbox™ and techniques including principle component analysis and self-organizing maps, they identified and organized projections according to slight conformational differences in the otherwise homogenous protein complexes.
The algorithms for pattern matching and single-particle reconstruction are computationally intensive. The scientists used Parallel Computing Toolbox™ to accelerate computation of these large datasets over a 64-node cluster.
Using algorithms developed with MathWorks tools, Max Planck researchers have already produced 3D images of several protein complexes in addition to the 26S proteasome. Current work includes optimizing and automating the workflow, using MathWorks tools to provide feedback to the microscope for the adaptive and optimal collection of data as required.
Results
Research time cut by years. “Researchers had been working for almost 10 years to build a 3D representation of the 26S proteasome,” says Korinek. “Using MathWorks tools we developed a workflow that produced the highest resolution structure available to date in less than two years.”
Development time cut from weeks to days. “With MATLAB we can develop a new algorithm, technique, or GUI in one or two days. The same effort would take at least a month in C++,” notes Korinek. “Because we have a single environment for our entire workflow, biologists can get started without training on multiple software packages.”
Workflow accelerated. “Reconstructing a 3D volume can take days on a single CPU. Using MATLAB and Parallel Computing Toolbox we deployed the algorithm to our cluster,” says Korinek. “This enabled us to speed up the process by 20 to 30 times and reduced a week’s job to an overnight run.”