Global Sensitivity Analysis with SimBiology
Learn about the Global Sensitivity Analysis (GSA) functionality in SimBiology®. You’ll discover:
- The differences between local and global sensitivity analysis and when it is appropriate to apply each method
- How Sobol indices and multiparametric GSA are calculated
- How to interpret the plots associated with Sobol and MPGSA
- How to choose your sample size for these GSA methods
You’ll also get an introduction to the concept of ‘Observables’ with respect to the model or data (for example, to calculate AUC) and how they can be used as outputs for a GSA.
Published: 5 Aug 2020
Welcome to the webinar. My name is Sietse Braakman. I'm an application engineer at MathWorks. Today, I will be talking about global sensitivity analysis with SimBiology.
The topics I wanted to discuss today are first to take you through some of the concepts in the global sensitivity analysis, to make sure that everyone understands and is on the same page with those concepts. Then I will explain how to perform global sensitivity analysis in SimBiology. We will be using two methods, the Sobol method and the multiparametric method. I will spend time in how to interpret these plots, as well as how to size your samples such that you get reliable results.
And we'll have time for a Q&A. There is a Q&A window part of the Webex that you can type your questions in during the during the meeting. Some of my colleagues, Fulden Buyukozturk and Jeremy Huard, are able to answer. And others, we might keep till the end for the Q&A session.
With that, let's get started. So we'll start with some of the concepts. Most of you might be familiar with the fact that there is local and global sensitivity analysis. So when I talk about local sensitivity analysis, I talk about an analysis around a single operating point in the parameter space. So you might have multiple parameters, and really, we're only performing this sensitivity analysis for a single set of those parameter values.
Global sensitivity analysis, on the other hand, is performed across a domain in your parameter space. So for every parameter, there is now an upper and a lower bound. And between those bounds, we explore how the model output is sensitive to those input parameters.
An important concept in sensitivity analysis is sampling. And so for local sensitivity analysis, this is mostly done one at a time. We are in that single operating point. We perturb one parameter and see how that affects the model output. We bring the parameter back to its original value and we'll perturb the next one, et cetera. And so it's one at a time. And as a result, you're unable to absorb observatory interactions between parameters from single analysis.
Global sensitivity analysis, on the other hand, is all at a time. So you take random samples from the parameter space to calculate the sensitivity index. As a result, you're able to observe interactions between the parameters. And then there are also multiple ways that you can sample that parameter space. In general, these samplings are uniform, but there are so-called low discrepancy sampling methods, such as Sobol, Latin hypercube, and Halton sequences, that you can use to perform the sampling.
And then lastly, there are multiple ways that you can calculate sensitivity indices. So for local sensitivity, we use a derivative or a ratio, how we look at the change in model output over the change in model input. For Sobol and for eFAST, which is Fourier-based method, you're using the variance. So you try to attribute variance to each parameter. And then there are distribution-based methods, such as the multiparametric global sensitivity analysis and correlation-based methods such as partial rank correlation coefficients.
The next thing to talk about is why should we use local or global sensitivity analysis. So here I have a very simple example, a one-compartment model with absorption ka, distribution volume Vd, and elimination-- parameterized enzymatic elimination so Vm and K max, the Vm and Km. Now, if I set every parameter to be 1 and I simulate this model for 10 hours, I get the following local sensitivity values. However, if I set ka to be 0.1, so I divide k by 10 and I do the same analysis, I get these results.
And so you can see that the results from the local sensitivity analysis strongly depend on that operating point in your parameter space. And so that's why it's advisable to use global sensitivity analysis if you don't know that operating point with much confidence. So in that case, global sensitivity analysis is most appropriate when you're exploring sensitivity across that parameter domain.
Still, there is a reason why you might want to use local sensitivity analysis, for example, for target identification. So if you do have a good idea of that calibration, and you have, for example, one calibration that represents a kidney-impaired patient, you might want to use local sensitivity analysis for target identification, such as Birgit Schoeberl and our colleagues have done in the paper below. And also, SimBiology in MATLAB use the local sensitivities to calculate the gradients in optimization algorithms.
Now, on the other hand, the global sensitivity analysis is more appropriate when you want to understand which model inputs drive the model response or a model-based decision metric if you don't know that operating point. And then you can use the results of that global sensitivity analysis also to inform parameter estimation strategy. So the parameters that the model is very sensitive to, you can-- most likely, you want to estimate, to make sure that you have a good understanding of what their value is and that your model is properly calibrated.
With that, I want to move on to Sobol global sensitivity analysis. This is one of the two methods that's being implemented in SimBiology, and I want to explain first how the Sobol index is calculated. So the idea is to apportion variance in the model output Y to the model inputs X.
And there are multiple indices you can calculate. The first order sensitivity index represents the individual contributions, so the individual variance that can be-- the variance that can be apportioned to each individual parameter. And it's calculated through this expression.
And in the denominator here, you see the unconditional variance. So assume we're simulating a one-compartment model, and we have-- we're sampling two parameters, the absorption coefficient ka and the clearance. Now, if I sample these parameters, I simulate each sample, I get this ensemble of simulations. And at every time point, I can calculate the variance of that ensemble, and that's what happens here in the denominator.
Now, in the numerator, you see the conditional variance. And that's the variance not due to Xi. Say, Xi is our parameter ka. So what we then do is we fix ka in one point. We still allow the clearance to vary, and we get the variance.
Now, we can do that for all values of ka, take the mean value, and then we can say that that is the variance that is due to not ka. And so 1 minus that whole value of that ratio gives us the variance due to ka. So that's the way that the first order Sobol index is calculated. So it's the ratio of the conditional variance over the unconditional variance, and then 1 minus that.
The other sensitivity index you can calculate is the total effect, and that shows interactions because it is the sum of all of the lower order effects that include your parameter of interest. So if your parameter or interest is number one or ka, it's the first order, the second order with each of the other parameters, the third order with each of the combination of parameters, et cetera. And that sum is basically-- there is an easier way to calculate that and that is through this expression, which I'm not going to go into much detail. But the idea of calculating it is similar to the first order sensitivity index.
OK, so that gives you an idea of how the Sobol indices are calculated and how that is different from a local sensitivity analysis, where you do the ratio between your change in model output over change in model input. So let's-- before we move to showing how you would do this in SimBiology, I just want to take you through the workflow that we're going to follow when we are moving to SimBiology.
So the first thing we're going to do is we're going to define the domain of interest in the parameter space. So I'm going to say, these are the parameters that I'm interested in incorporating in my global sensitivity analysis, and for each of them, I need to decide what the lower and upper bound is. Then that defines my parameter space, and I can sample that. I can simulate each sample. And then once all my simulations are complete, I can calculate the sensitivity measures.
I'm going to demonstrate this using a model by Sergey Aksenov and his colleagues. This is a model that describes lesinurad and febuxostat, which are two approved drugs to treat gout. And the model looks like this. So we have a two-compartment model for the lesinurad at the top and a two-compartment model for the febuxostat at the bottom. And you can see that the febuxostat, the central concentration, has an effect on the production of serum uric acid, whereas the lesinurad increases the glomerular filtration and thereby basically increases the clearance of uric acid to avoid accumulation of it.
What I'm going to do today, I'm going to choose four parameters from this model, particular the PD part of the model, to make sure that we have-- that we explore these four parameters and see to what extent they have an effect on the output, which I choose to be serum uric acid. So the four parameter are k1, fc50, fmax, and e0. And of course, I could take more parameters, but then we're going to spend a lot of time simulating and that's-- for the purpose of today, it's not helpful.
All right, there are two ways that you can perform this analysis. You can either perform it entirely programmatically, so you can write your own script, or you can use an app to perform the analysis. So the app, you can download from-- if you go to MATLAB, to the Home tab, and you click here on the Add-On Explorer, and you search for global sensitivity analysis SimBiology, you can download this app here and install it directly onto your machine.
If you can't access this for some reason, note that the functions themselves are built in and installed as of 2020a-- SimBiology 2020a. So the two functions are sbiosobol and sbiompgsa, and the app is basically a user interface to that-- to those two functions. So what we do is-- we will use the app today, just so it's easier for you to follow what I'm doing. But at the end of the presentation, I will share my code and the model with you, such that you can also try it out using code.
In order to run the app, what I need to do is I need to move this model to MATLAB. So I can export this model to the workspace, to my MATLAB workspace. For example, calling it m1. And then in MATLAB, I can pull out the doses. So in this case, I want to use dose one and two. I've already done all of that and started up the app, but you call the app like this, startGlobalSensi tivityAnalysisApp, and then you pass in the model and the doses. And that starts up the app, and it looks like this.
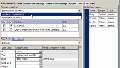
So let's walk through this step by step. The first thing, as I said, that we need to do is we need to define which parameters we are interested in. So I can go here. That basically opens up a separate window with all of the objects in my model, so the compartments, parameters, initial conditions for the species, et cetera, that I can use as inputs to my global sensitivity analysis. And I can sort those.
And so what I'm interested now in is selecting these four parameters that are of interest to me, e0, fc50, fmax, and k1. And now I can choose values, upper and lower bound values, for each of those. So I've already done that, and I've ended up basically taking the model value, the standard value of the parameter in the model, then divide by 1 and 1/2 and times 1 and 1/2, primarily because it's easier to always bound it by 1. So I wanted to make sure that all of the parameters had a similar width in terms of order of magnitude spread between the upper and lower bound.
And you can just edit this and write 0.5, and then change the parameter values. So then when you're done, you've selected all your parameters and input all the upper and lower bounds, we move on. Here, you can select the number of samples. By default, this is 1,000, and I'm going to keep it that way.
And then you need to define the output times, and this is basically MATLAB code. So you can use linspace, but you can also just write your own time vector here. So I'm going to use the vector that goes from 0 to 180 in steps of 0.5 hours.
Then there are a few dropdown menus here. You can choose different sampling methods. As I said, Sobol, Halton, and Latin hypercube, those are low discrepancy sampling methods, which-- I recommend using one of those three because it's more efficient than using just a standard random uniform distribution.
The output times are reported after the simulation is done, and they might not coincide with the steps that ODE solver has taken. So that's why you can choose an interpolator. I would just recommend using the default value here.
And then there are different ways that you can speed up this global sensitivity analysis. So first of all, you can parallelize the simulations. I will be using a parallel pull with four workers here, on my core i7 laptop with four cores. And we will also accelerate the model, so that that compiles the model to seek code in order to speed up the simulations. And then we can talk about that later.
So that's basically the input part of the global sensitivity analysis that we've set up. Now, the next thing we can do is we can define what the output of interest is for us. Well, and for this model for gout, the output of interest is the serum uric acid levels. So in here, you can select whichever output you're interested in. If you're just interested in looking at pKa for lesinurad, you could use this-- the central lesinurad concentration.
But in this case, we're very interested in the clinically-relevant output of the model, which is the serum uric acid. So I just select that and click Done. And then we can basically start simulating the model. And I'll talk about this more in a bit. But basically, the combination of having four parameters and drawing 1,000 samples means that we need to do 1,000 times 4 plus 2, so 6,000 simulations.
So I'm going to go ahead and start that now, and in the meantime, I'm going go back to the slides to discuss another new feature in SimBiology that is relevant for this particular case. And those are called observables. So the idea behind observables is that they supersede and expend calculate statistics functionality.
So you might be familiar with the calculate statistics functionality, where you simulate your model in the Task Editor in 2019a and prior, and you were able to calculate, for example, cmax or something. It always had to be a scalar value. The observables, built on top of that, and they don't necessarily have to result in scalar, but they can also be time-based. And so time-based might be relevant if, for example, you have different species that you want to add up to get, for example, the total tumor volume, or total drug concentration, or something like that.
And the idea is the observables, you can apply to your simulation data. So say, you have done 100 simulations, and you just the AUC from each of those 100 simulations. So you have a 100 by 1 SimData array, and you can then add an observable to that SimData array that just says, OK, give me the AUC which equals trapz time, central.drug_central, and then it will just give you 100 AUCs, basically. And so that will make your life a lot easier. You can do that in one line of code.
Now, some of you might realize that this could also be achieved by using repeated assignments. So the difference between repeated assignment and observables are that the observables are calculated after the ODEs are solved. So they're not part of the system of equations. And so observables cannot be variables that the system depends on. If your model or your system depends on that tumor volume, for example, to change the compartment tumor volume, then it needs to be a repeated assignment. But if nothing in the model depends on it, it's better to use an observable because it's less computationally expensive.
The observables can be applied to both your model and to the SimData object. If you add an observable to a model and you simulate the model, the observable is automatically calculated as well. And the SimData, if you add the observable to the SimData, then it's calculated and returns, for example, that AUC.
OK, so I hope that makes sense for the observables. So let's go back to SimBiology. It looks like it's done now, and we have our results here. Last time, I did this earlier today. It took about 90 seconds to simulate these 6,000 simulations. So-- OK.
So here we have the results. We have in blue the first order sensitivities, Sobol sensitivities, and in red the total order sensitivities. So what you can see is that e0 is clearly the most important parameter in the model, probably followed by k1, fmax, and then fc50.
Now, if there were no interactions, these should all add up to 1 at any moment in time or around about 1. You can see that that's not quite the case. So there is-- here is a fraction of unexplained variance that is not 0. And so this gives you an indicator that there is some interaction between your parameters.
There are other reasons this could be non-zero, which is, you might have some numerical drift or something in your simulation. But the most common explanation would be that there are interactions. And you can see that there are interactions by comparing the total order values, for example, here, to the first order values.
And so if we compare these two, you can see that this one is higher than this one, and that's what you expect. You expect the total order to be either the same or higher than the first order because the total order is the sum of the first order, all of the second, third, et cetera, orders. And so by comparing these two, you can see which parameters show most interaction.
OK, so this gives us an idea of the global sensitivities over time. The output that we took was serum uric acid, which is a continuous variable throughout-- for the model that changes over time. And the waves that you see here are the different dosing events. And so what we can see here is that e0 here looks like it's more important at the earlier stages of the simulation than at the later stages.
And the other thing, of course, we could have done was we could have used an actual observable, scalar observable, like AUC or the minimum value of the serum uric acid as our model output. And then of course, we wouldn't have had a time course, but we would have had a single number for each scalar value, for each of the first and total order indices.
Another thing you can plot, and that's generally good to do, is this sort of sanity check-- is you can plot the data. So if you plot the data, you can see the results here of all the simulations with the 90 percentile region in blue, and some of the individual traces dotted here. And in red, you see the mean simulation value for all of the samples, the 1,000 samples that we took.
OK, so with that, we're going to move on to the multiparametric global sensitivity analysis. And the idea behind the multiparametric global sensitivity analysis is that you use a classifier to analyze the relative importance of parameters, and that classifier is a model-based decision metric based on the model outputs, but it has to result in a true or false outcome. And so as a result, there has to be an inequality. So the effect-- for example, the classifier can be, my pharmacodynamic effect is larger than 70% or the final concentration is larger than the mean of the final concentration.
And so that way, you can basically classify your simulations between, do they meet that classifier? Are they indeed larger than 70%? Or do they fail? Are they rejected?
You can use a combination of these. Like, you can create a single, the effect has to be larger and the final concentration has to be larger than that. Or you can also just perform the multiparametric global sensitivity with multiple outputs, with multiple classifiers, and see which ones are relevant for you.
So here's an example of how-- what it looks like. Say you have a model and there are two parameters that you're varying, kel and IC50. Then you simulate the model for these two values, and you see whether the effect is larger than 70. And if so, then you say yes, and otherwise, you say no. And so that way, we get a set of simulations that are either accepted or rejected.
And from that, we can calculate an empirical cumulative distribution function. So if they are-- and we do that for both the accepted and for the rejected sample. So you can see an example here. For k1, this is the case. So at low values of k1, it looks like more of the samples are accepted, whereas at higher values, more of the samples are rejected.
And what we can then do is we can calculate the maximal distance. So that's the maximum distance between the two functions vertically, basically. And we can use a Kolmogorov-Smirnov test to see whether these distributions are statistically significantly different. And so the advantages of doing this over a Sobol is that you actually get an answer, like, is there a significantly different result. And you can, of course, choose that metric, that classifier to be relevant for your case.
Now, there's one thing I haven't touched on and that's the threshold. That threshold, for example, 70%, that should be about the halfway mark on your simulation. So about 1/2 of the simulations should pass and 1/2 of those simulations should fail, in order to be able to construct those two cumulative distribution functions.
Because if more of them failed than pass, then you're going to get fewer passed ones. And you're going to get a very jagged CDF, and then your Kolmogorov-Smirnov test is not going to be reliable. So what you can actually do is, you can use this simulation here, that plot of all your simulations, and for example, use that red line to come up with a threshold.
OK, so how do we bring this in practice? Well, it's actually very simple. We can go back here, and we can actually reuse the simulations. And we already did the Monte Carlo simulation. So we can just reuse those simulations. The only thing I need to define is my classifier.
And so I can type that classifier in. And I can just-- I don't have to run the simulations anymore. I can just compute the multiparametric global sensitivity analysis.
And so here, you see the results from the multiparametric global sensitivity analysis. And you see that k1 and fmax are-- now there is a significant difference between the two. For e0, they are miles apart. And for fc50, they're very close together.
And you can compare this to the histogram as well. The cumulative distribution function and the histogram are related. And so you can see, for example, for e0, that most of the accepted samples occur at lower values of e0. And that's why you see this blue line rise. Whereas the rejected samples only occur at higher values, and that's why the red line only comes of 0, like rises above 0, at these higher values of e0. So that's how the histogram and the eCDF are related.
And so what we're looking at with this Kolmogorov-Smirnov test is how different are these histograms or how different are the-- really, the Kolmogorov-Smirnov test looks at how different are these cumulative distribution functions. And how we can do that is we can plot-- called bar. And that plots are-- that calculates the ks statistics, which you see here in blue, and the p-value of whether it's statistically significantly different.
So then you see that for fmax, e0, and k1, the effect is significant. So the two distributions are significantly different. Whereas for fc50, it doesn't reach the threshold of p is less than 0.05.
OK, so that concludes the multiparametric global sensitivity analysis. There is one more topic I want to discuss and that is the number of iterations. And so in order to perform this global sensitivity analysis, you have to calculate-- you have to perform a great deal of simulations. It's computationally expensive. But also, in order to get reliable results, you need to have-- you can't undersample. So you know, what is a good number of samples to have?
So if we assume that N is the number of samples that we draw from our parameter input space and P is the number of input parameters, so that's the dimensionality of the parameter input space, then we can say whichever is larger. So N should be larger than 2 to the power of P. That would be the absolute bare minimum.
If I have two parameters, then I take four samples. That only allows me to cover the corners of my input space. If I have three parameters, it's eight, so that's the corners of the cube. So really, what you want is a higher base number. So like, 3 to the power of P or 4 to the power of P.
And then you can see that you know as P increases above like, 15, that you're looking at a very large number of simulations. And in general, for-- if you have-- if you're performing this with less than five or less parameters, I would recommend getting 1,000 to 5,000 simulations, like what I did here. I did 1,000 simulations for those four parameters.
So try that out. There are a few ways that you can see that you're actually undersampling, and that is if your Sobol results are falling below 0. So this is just above 0, but if this were negative, then I would be worried about my-- about undersampling-- or if they are above the above 1.
Because the Sobol index is always trying to see what percentage of the variance can be-- of the total variance can be attributed to the single parameter. It should always be between 0 and 1. So that's a surefire symptom that something is wrong, and you should increase the number of samples.
Another one, of course, is that if you rerun the analysis, you're getting meaningfully different results, but you might not have the computational resources to try it out multiple times. And lastly, if you have-- for MPGSA, if you have a jagged or a staircase shape-- I touched on this a little bit earlier-- then you might either have a poor choice of your threshold-- that threshold might not be placed such that about 1/2 are rejected and 1/2 are accepted-- but it can also be due to insufficient sample size. And you need to know that if you have that jagged staircase, you should not trust the results from your MPGSA.
Lastly, because we do-- we have to do so many simulations and because we have to simulate all of the samples before we can calculate the sensitivity measures, then all of the simulation results need to be held in memory in order to calculate the sensitivity indices. And so if you can minimize the memory footprint of your simulation, you can probably perform more samples. You can simulate more samples.
So how can you minimize the memory footprint? Well, first of all, you can reduce the number of output. So if you only have a single output, like the final concentration or final effect, that will greatly minimize the number of outputs. And also limiting the number of logged states. So make sure that there are no-- that you're not logging all of your species, et cetera. And then lastly, of course, you can use an observable in order to minimize that footprint as well because then you can basically boil down an entire simulation into a single scalar using that observable, and that will allow you to take more, to run a larger global sensitivity analysis.
OK, so in summary, we talked about what do we need to do in order to perform this sensitivity analysis. We need to define that domain, what are the upper and lower bounds for each parameter. Then we can sample that domain. For each sample, we can simulate the model. And then once we've simulated all of the samples, we get that ensemble of simulations. From there, we can calculate the sensitivity measure.
So in summary, some of the things you should think about, which parameters do you want to include, what is the range for each of those parameters. And that range can be physiologically defined, or it can be that you have found different values in literature, and you take the lowest value you found and the highest value you found. Then you're going to sample. You need to choose a sampling method. All of these samples have to be uniform in order for assumptions underlying the Sobol indices calculation to not be violated.
So the Latin hypercube, this Sobol sequence, and the Halton sequence, they're all uniform sampling methods. But they have an advantage of the random uniform sampling, in that they require fewer samples to be taken, while still covering the same input space. So I recommend using Latin hypercube, Sobol, or Halton for your sampling. And then you also need to choose the number of samples.
Then you're going to simulate the model, so you need to make sure you add the doses. If you don't add a dose, you're not perturbing the model, and you're not going to see any results. The variants need to be right. You need to define what are time points, what the model output of interest is, what your classifier is, et cetera, and think here again about your memory footprint.
And then lastly, we can talk about the-- well, sbiosobol and sbiompgsa, the two functions that are basically working under the hood of that app that I showed you, they will calculate the first order and total order Sobol indices and the eCDFs, and perform the K-S tests on all of the simulations that you then run.
Featured Product