Visualize NGS Data Using Genomics Viewer App
The Genomics Viewer app lets you view and explore integrated genomic data with an embedded version of the Integrative Genomics Viewer (IGV) [1][2]. The genomic data include NGS read alignments, genome variants, and segmented copy number data.
The first part of this example gives a brief overview of the app and supported file formats. The second part of the example explores a single nucleotide variation in the cytochrome p450 gene (CYP2C19).
Open the App
At the command line, type genomicsViewer. Alternatively, click the
app icon on the Apps tab. The app requires an internet connection.
By default, the app loads Human (GRCh38/hg38) as the reference sequence and Refseq Genes as the annotation file. There are two main panels in the app. The left panel is the Tracks panel and the right panel is the embedded IGV web application. The Tracks panel is a read-only area displaying the track names, source file names, and track types. The Tracks panel updates accordingly as you configure the tracks in the embedded IGV app.
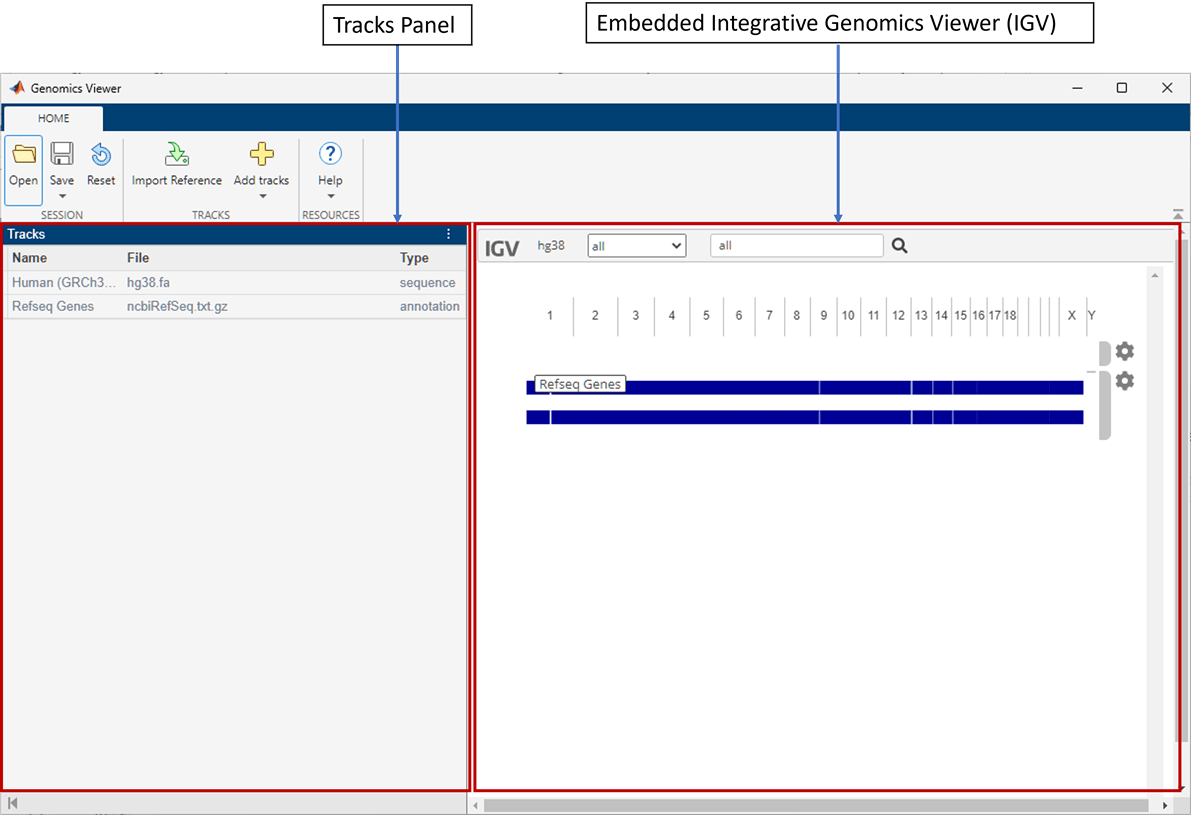
The Reset button restores the app to the default view with two
tracks (HG38 with Refseq Genes) and removes any other existing tracks. Before resetting, you
can save the current view as a session (.json) file and restore it later.
Add Tracks by Importing Data
You can import data from local files or specify URLs. If you are specifying a URL, the
URL must start with http, https, or
gs. Other file transfer protocols, such as ftp, are
not supported. For a list of supported data file formats, see https://igvteam.github.io/igv-webapp/fileFormats.html.
Import Reference Sequence
You can import a single reference sequence. The reference sequence must be in a FASTA file. Select Import Reference on the Home tab. You can also import a corresponding cytoband file that contains cytogenetic G-banding data.
Import Sequence Read Alignment Data
You can import multiple data sets of sequence read alignment data. The alignment data
must be a BAM or CRAM file. It is not required that you have the corresponding index file
(.BAI or .CRAI) in the same location as your BAM
or CRAM file. However, the absence of the index file will make the app slower.
You can add read alignment files using Add tracks from file and Add tracks from URL options from the Add tracks button.
Import Feature Annotations and Other Genomic Data
You can import multiple sets of feature annotations from several files that contain
data for a single reference sequence. The supported annotation files are:
.BED, .GFF, .GFF3, and
.GTF.
You can also import structural variants (.VCF) and visualize genetic alterations, such as insertions and deletions.
You can view segmented copy number data (.SEG) and quantitative
genomic data (.WIG, .BIGWIG, and
.BEDGRAPH), such as ChIP peaks and alignment coverage.
You can add annotation and genomic data files using Add tracks from file and Add tracks from URL options from the Add tracks button.
Visualize Single Nucleotide Variation in Cytochrome P450
The CYP2C19 gene is a member of the cytochrome P450
gene family. Enzymes produced from cytochrome P450 genes are involved in the metabolism of
various molecules and chemicals within cells. The CYP2C19 enzyme plays a role in the
metabolizing of at least 10 percent of commonly prescribed drugs [3]. Polymorphisms in the
cytochrome p450 family may cause adverse drug responses in individuals. One example of
single nucleotide variation is rs4986893 at position
chr10:94,780,653 where G is replaced by
A. This allelic variant is also known as CYP2C19*3.
The following steps show how to visualize such variation in the app using both low coverage
and high coverage data.
Load Session File
For the purposes of this example, start with a session file (rs4986893.json) that has some preloaded tracks. After downloading the file, load it in the app. Click Open and select rs4986893.json.
Explore Low Coverage Data
The session contains three tracks:
Human (GRCh38/hg38) as a reference
NA18564 as low coverage alignment data
Refseq Genes
The low coverage alignment data comes from a female Han Chinese from Beijing, China. The sample ID is NA18564 and the sample has been identified with the CYP2C19*3 mutation [4].
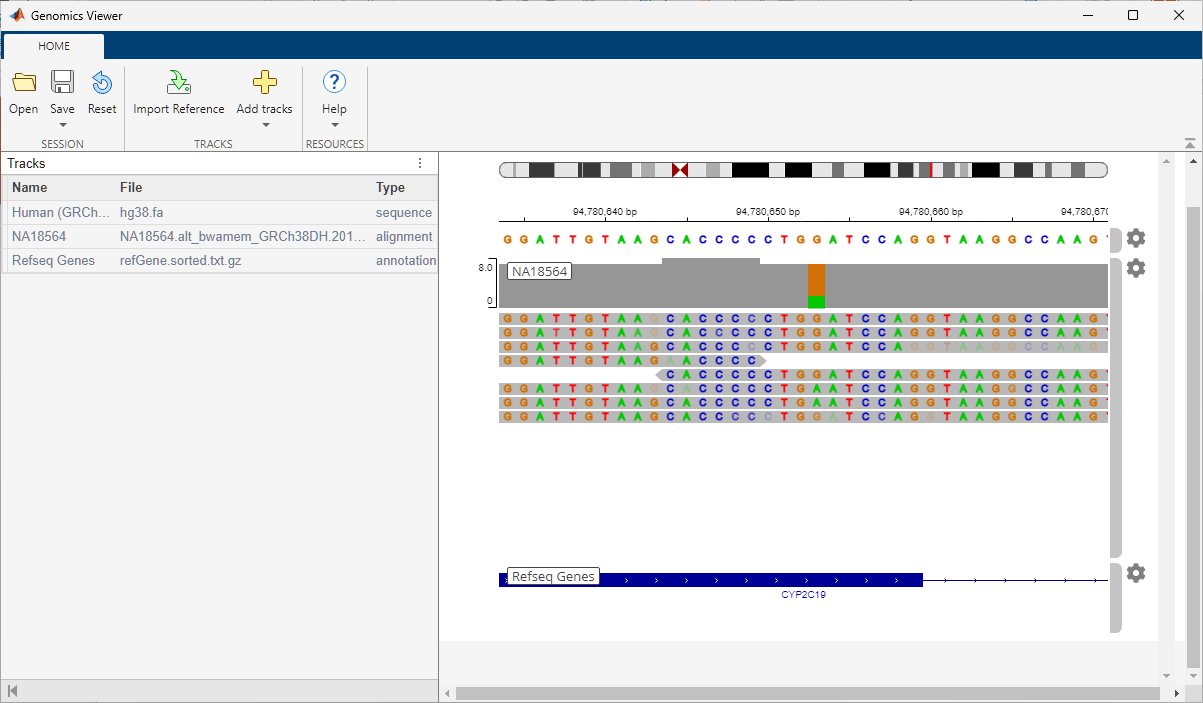
The alignment data has been centered around the location of the mutation on the CYP2C19 gene.
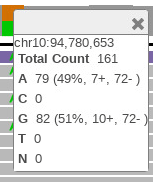
Click the orange bar in the coverage area to look at the position and allele distribution information.
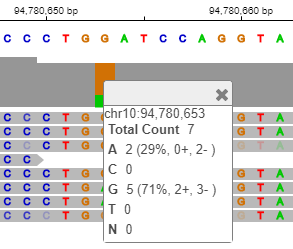
It shows that 71% of the reads have G while 29% have A at the location chr10:94,780,653. This data is a low coverage data and may not show all the occurrences of this mutation. A high coverage data will be explored later in the example.
Close the data tip window.
You can customize the various aspects of the data display in the app. For example, you can change the track height to make more room for later tracks. Click the second gear icon. Select
Set track height. Enter 200.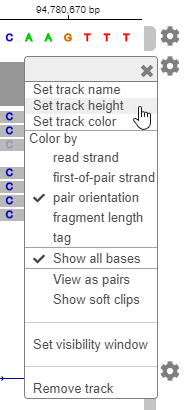
For details on the embedded IGV app and its available options, visit here.
Explore High Coverage Data
You can look at the high coverage data from the same sample to see the occurrences of this mutation.
Go to The International Genome Sample Resource website.
Search for the sample NA18564.
Download the Exome alignment file that is in the
.CRAMformat.Also download the corresponding index file that is in the
.CRAIformat. Save the file in the same location as the source.CRAMfile.Click the (+) icon on the Home tab. Select the downloaded
.CRAMfile and click Open.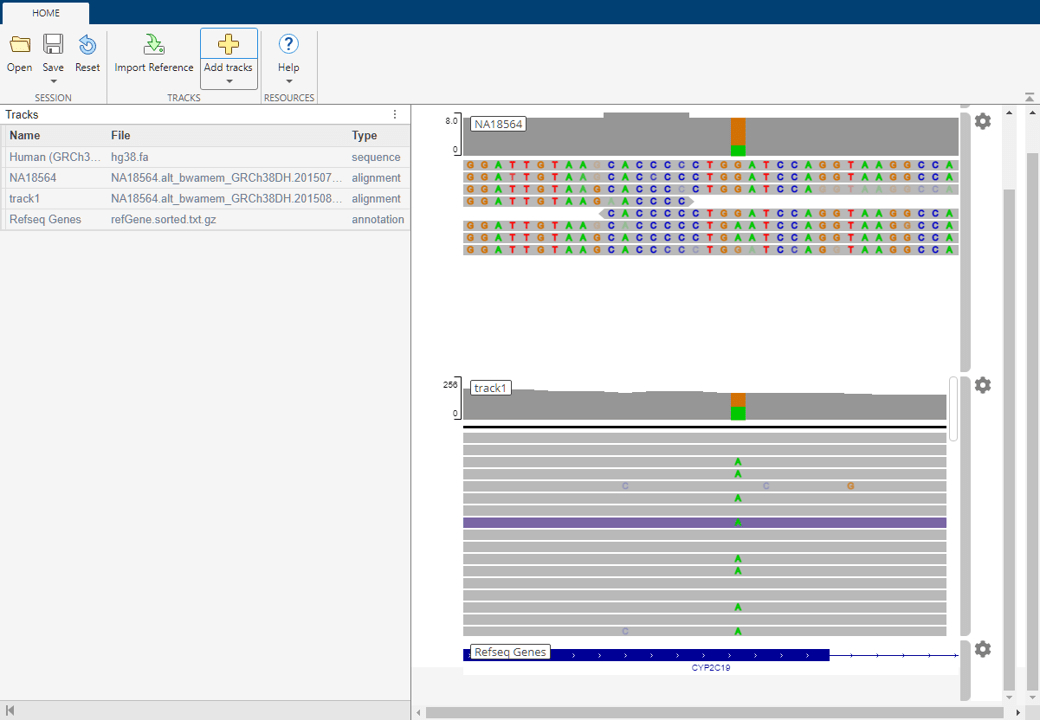
The high coverage data appears as a track. You can now see many occurrences of the mutation in several reads.
Click the orange bar in the coverage area to see the allele distribution.
References
[1] Robinson, J., H. Thorvaldsdóttir, W. Winckler, M. Guttman, E. Lander, G. Getz, J. Mesirov. 2011. Integrative Genomics Viewer. Nature Biotechnology. 29:24–26.
[2] Thorvaldsdóttir, H., J. Robinson, J. Mesirov. 2013. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Briefings in Bioinformatics. 14:178–192.
See Also
Genomics Viewer | bioinfo.pipeline.block.GenomicsViewer | Sequence Alignment