eval
Description
outStruct = eval(blockObj,inStruct)blockObj using the input structure
inStruct and returns the block outputs in the output structure
outStruct.
Note
It is recommended that you use the
runmethod of a block instead ofevalbecause therunmethod performs additional error checks and ensures that the block inputs and outputs are satisfied and that theevalmethod accepts and returns a scalar structure, which is a requirement to run the block as part of a pipeline.For all block objects, the
runmethod of a block calls theevalmethod.runis a sealed method that performs some validation on the inputs and outputs foreval. Theevalmethod is a customized evaluation method of a block with additional validation on the inputs and outputs and is sealed. To implement a custom block behavior, create a block subclass and write your ownevalfunction as described in this example.Do not call
evaldirectly unless you are prototyping your own block subclass and implementing your block tasks in theevalmethod.When you implement your custom
evalmethod for a block, ensure that the method does not modify any properties of the block so that the pipeline can skip execution of the block when the inputs are the same as a previous run.
Examples
For most pipelines, using a combination of built-in blocks and UserFunction blocks is sufficient and recommended. However, if a block requires more complexity, such as additional configuration options or methods, you can create a custom block by subclassing the bioinfo.pipeline.Block object to implement additional capabilities.
Subclasses of the Block object must:
Define their
InputsandOutputsproperties.Define the
evalmethod.
For instance, the following lines of code defines a custom block object named Seqlinkage which is defined as a subclass of bioinfo.pipeline.Block. For illustration purposes, the Seqlinkage block uses the seqlinkage function, which constructs a phylogenetic tree from pairwise distances as the underlying function. The function accepts a matrix or vector of pairwise distances and a distance method to use.
classdef Seqlinkage < bioinfo.pipeline.Block % Define the block properties properties % Define the Method property that accepts two distance methods. % For details on these methods, enter the following command at % the command line: doc seqlinkage Method {mustBeMember(Method, ["average", "weighted"])} = "average"; end methods % Define inputs and outputs. function block = Seqlinkage() import bioinfo.pipeline.Input import bioinfo.pipeline.Output % Define the Inputs and Outputs property of the object. % Name the first input port asd Distances and as a required % input that takes in a matrix or vector of pairwise distances. block.Inputs.Distances = Input('Required',true); % Name the second input port as Names and as an optional % input that takes in a list of names for nodes. block.Inputs.Names = Input('Required',false); % Name the output port Phytree. block.Outputs.Phytree = Output; end % Define custom evaluation method for the block. function outStruct = eval(obj, inStruct) % If the optional input (a list of node names) is passed in if isfield(inStruct,'Names') % Call the seqlinkage function with three inputs and save % the returned phytree object as output. outStruct.Phytree = seqlinkage(inStruct.Distances,obj.Method,inStruct.Names); else % Call seqlinkage with two inputs. outStruct.Phytree = seqlinkage(inStruct.Distances,obj.Method); end end end end
You can save such a class definition to a separate MATLAB® program file. For this example, the above Seqlinkage class definition has already been saved and provided as Seqlinkage.m. Note that the definition file must be in the current working folder or MATLAB search path before you can use the block object in your pipeline.
Next, you can define a pipeline that uses the Seqlinkage block as follows to build a phylogenetic tree from pairwise distances.
import bioinfo.pipeline.Pipeline import bioinfo.pipeline.block.* P = Pipeline;
Create three blocks needed for the workflow.
fastaReadBlock = UserFunction("fastaread",RequiredArguments="inputFile",OutputArguments="sequences"); seqpdistBlock = UserFunction("seqpdist",RequiredArguments="inputSequences",OutputArguments="pairwiseDistances"); seqlinkageBlock = Seqlinkage;
Set the input port of fastaReadBlock to a FASTA file containing amino acid sequences.
fastaReadBlock.Inputs.inputFile.Value = which("pf00002.fa");Add the blocks to the pipeline.
addBlock(P,[fastaReadBlock,seqpdistBlock,seqlinkageBlock],["fastaread","seqpdist","seqlinkage"]);
Connect the first two blocks.
connect(P,"fastaread","seqpdist",["sequences","inputSequences"]);
Connect seqpdistBlock to seqlinkageBlock. Specifically, connect the output port "pairwiseDistances" of seqpdistBlock to one of the input ports of the seqlinkageBlock "Distances", as defined in the Seqlinkage.m. Note that Distances is a required input port that must have its value set. One of the valid ways is to connect to another port. For other ways to set input ports, see Satisfy Input Ports.
connect(P,"seqpdist","seqlinkage",["pairwiseDistances","Distances"]);
Connect the output port "sequences" of the fastaread block to the "Names" optional input port of seqlinkageBlock to label the leave nodes of the output phylogenetic tree.
connect(P,"fastaread","seqlinkage",["sequences","Names"]);
Optionally, you can also pass in a list of node names as the value of the optional input port "Names" by setting the property seqlinkageBlock.Inputs.Names.Value.
Before you run the pipeline, ensure that the folder of the class definition file is on the MATLAB search path. The reason is because each pipeline block runs in its own folder, and the external class definition files or function files will not be detected by the pipeline unless they are on the path.
% Update the following addpath call to specify the path to your class definition file. For instance, if Seqlinkage.m is located in C:\Examples\SubclassExample\, use the following: addpath("C:\Examples\SubclassExample\");
Run the pipeline.
run(P);
Get the result from seqlinkageBlock, which contains a phylogenetic tree object.
linkageResult = fetchResults(P,seqlinkageBlock)
linkageResult = struct with fields:
Phytree: [1×1 phytree]
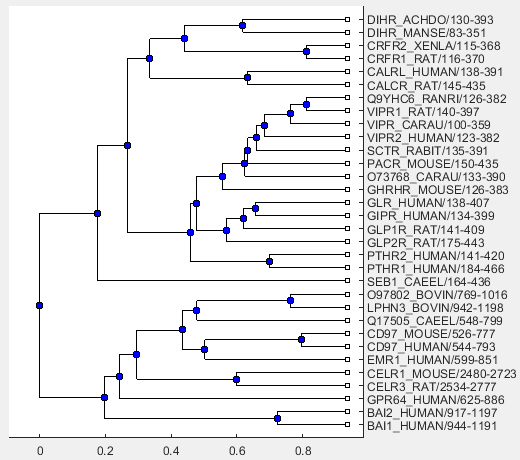
Use the following command to visualize the phylogenetic tree.
view(linkageResult.Phytree)
Input Arguments
Output Arguments
Version History
Introduced in R2023a
See Also
compile | bioinfo.pipeline.Block | bioinfo.pipeline.Pipeline