bioinfo.pipeline.Block
Description
The bioinfo.pipeline.Block object is used in a bioinformatics
pipeline to perform a unit of work, such as aligning reads to a reference, that is necessary
to achieve the final goal of an analysis pipeline.
Bioinformatics Toolbox™ provides built-in blocks to accomplish some bioinformatics-specific tasks. For
instance, you can use the SeqTrim block to trim genomic reads and
Bowtie2 block to align reads to a reference genome.
In addition to built-in blocks, you can also convert any existing MATLAB® function into a block by using a UserFunction block and use it
in your pipeline.
You can implement most pipelines and analysis workflows by using a combination of
UserFunction blocks and built-in blocks. However, if you are a developer or
advanced user who needs to customize a block behavior beyond those of the built-in blocks and
UserFunction blocks, you can create your own block object as a subclass of
the bioinfo.pipeline.Block class. For details, see Subclass Pipeline Block.
Creation
To create one of the built-in blocks, use
bioinfo.pipeline.block.BlockName, where
BlockName is the name of the built-in block. For example, to create a
SamSort block, enter bioinfo.pipeline.block.SamSort.
Similarly, to create a UserFunction block, use
bioinfo.pipeline.block.UserFunction.
Tip
To see a list of built-in blocks at the MATLAB command line, enter
bioinfo.pipeline.block.
Properties
Object Functions
compile | Perform block-specific additional checks and validations |
copy | Copy array of handle objects |
emptyInputs | Create input structure for use with run method |
eval | Evaluate block object |
run | Run block object |
Examples
Import the Pipeline and block objects needed for the example.
import bioinfo.pipeline.Pipeline import bioinfo.pipeline.block.*
Create a pipeline.
qcpipeline = Pipeline;
Select an input FASTQ file using a FileChooser block.
fastqfile = FileChooser(which("SRR005164_1_50.fastq"));Create a SeqFilter block.
sequencefilter = SeqFilter;
Define the filtering threshold value. Specifically, filter out sequences with a total of more than 10 low-quality bases, where a base is considered a low-quality base if its quality score is less than 20.
sequencefilter.Options.Threshold = [10 20];
Add the blocks to the pipeline.
addBlock(qcpipeline,[fastqfile,sequencefilter]);
Connect the output of the first block to the input of the second block. To do so, you need to first check the input and output port names of the corresponding blocks.
View the Outputs (port of the first block) and Inputs (port of the second block).
fastqfile.Outputs
ans = struct with fields:
Files: [1×1 bioinfo.pipeline.Output]
sequencefilter.Inputs
ans = struct with fields:
FASTQFiles: [1×1 bioinfo.pipeline.Input]
Connect the Files output port of the fastqfile block to the FASTQFiles port of sequencefilter block.
connect(qcpipeline,fastqfile,sequencefilter,["Files","FASTQFiles"]);
Next, create a UserFunction block that calls the seqqcplot function to plot the quality data of the filtered sequence data. In this case, inputFile is the required argument for the seqqcplot function. The required argument name can be anything as long as it is a valid variable name.
qcplot = UserFunction("seqqcplot",RequiredArguments="inputFile",OutputArguments="figureHandle");
Alternatively, you can also use dot notation to set up your UserFunction block.
qcplot = UserFunction; qcplot.RequiredArguments = "inputFile"; qcplot.Function = "seqqcplot"; qcplot.OutputArguments = "figureHandle";
Add the block.
addBlock(qcpipeline,qcplot);
Check the port names of sequencefilter block and qcplot block.
sequencefilter.Outputs
ans = struct with fields:
FilteredFASTQFiles: [1×1 bioinfo.pipeline.Output]
NumFilteredIn: [1×1 bioinfo.pipeline.Output]
NumFilteredOut: [1×1 bioinfo.pipeline.Output]
qcplot.Inputs
ans = struct with fields:
inputFile: [1×1 bioinfo.pipeline.Input]
Connect the FilteredFASTQFiles port of the sequencefilter block to the inputFile port of the qcplot block.
connect(qcpipeline,sequencefilter,qcplot,["FilteredFASTQFiles","inputFile"]);
Run the pipeline to plot the sequence quality data.
run(qcpipeline);
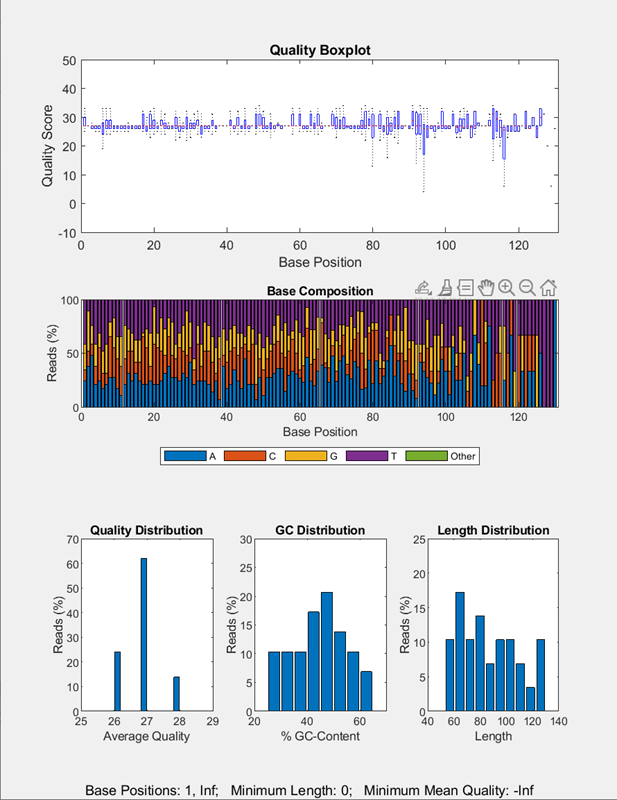
More About
For most pipelines, using a combination of built-in blocks and UserFunction blocks is sufficient and recommended. However, if a block requires more complexity, such as additional configuration options or methods, you can create a custom block by subclassing the bioinfo.pipeline.Block object to implement additional capabilities.
Subclasses of the Block object must:
Define their
InputsandOutputsproperties.Define the
evalmethod.
For instance, the following lines of code defines a custom block object named Seqlinkage which is defined as a subclass of bioinfo.pipeline.Block. For illustration purposes, the Seqlinkage block uses the seqlinkage function, which constructs a phylogenetic tree from pairwise distances as the underlying function. The function accepts a matrix or vector of pairwise distances and a distance method to use.
classdef Seqlinkage < bioinfo.pipeline.Block % Define the block properties properties % Define the Method property that accepts two distance methods. % For details on these methods, enter the following command at % the command line: doc seqlinkage Method {mustBeMember(Method, ["average", "weighted"])} = "average"; end methods % Define inputs and outputs. function block = Seqlinkage() import bioinfo.pipeline.Input import bioinfo.pipeline.Output % Define the Inputs and Outputs property of the object. % Name the first input port asd Distances and as a required % input that takes in a matrix or vector of pairwise distances. block.Inputs.Distances = Input('Required',true); % Name the second input port as Names and as an optional % input that takes in a list of names for nodes. block.Inputs.Names = Input('Required',false); % Name the output port Phytree. block.Outputs.Phytree = Output; end % Define custom evaluation method for the block. function outStruct = eval(obj, inStruct) % If the optional input (a list of node names) is passed in if isfield(inStruct,'Names') % Call the seqlinkage function with three inputs and save % the returned phytree object as output. outStruct.Phytree = seqlinkage(inStruct.Distances,obj.Method,inStruct.Names); else % Call seqlinkage with two inputs. outStruct.Phytree = seqlinkage(inStruct.Distances,obj.Method); end end end end
You can save such a class definition to a separate MATLAB® program file. For this example, the above Seqlinkage class definition has already been saved and provided as Seqlinkage.m. Note that the definition file must be in the current working folder or MATLAB search path before you can use the block object in your pipeline.
Next, you can define a pipeline that uses the Seqlinkage block as follows to build a phylogenetic tree from pairwise distances.
import bioinfo.pipeline.Pipeline import bioinfo.pipeline.block.* P = Pipeline;
Create three blocks needed for the workflow.
fastaReadBlock = UserFunction("fastaread",RequiredArguments="inputFile",OutputArguments="sequences"); seqpdistBlock = UserFunction("seqpdist",RequiredArguments="inputSequences",OutputArguments="pairwiseDistances"); seqlinkageBlock = Seqlinkage;
Set the input port of fastaReadBlock to a FASTA file containing amino acid sequences.
fastaReadBlock.Inputs.inputFile.Value = which("pf00002.fa");Add the blocks to the pipeline.
addBlock(P,[fastaReadBlock,seqpdistBlock,seqlinkageBlock],["fastaread","seqpdist","seqlinkage"]);
Connect the first two blocks.
connect(P,"fastaread","seqpdist",["sequences","inputSequences"]);
Connect seqpdistBlock to seqlinkageBlock. Specifically, connect the output port "pairwiseDistances" of seqpdistBlock to one of the input ports of the seqlinkageBlock "Distances", as defined in the Seqlinkage.m. Note that Distances is a required input port that must have its value set. One of the valid ways is to connect to another port. For other ways to set input ports, see Satisfy Input Ports.
connect(P,"seqpdist","seqlinkage",["pairwiseDistances","Distances"]);
Connect the output port "sequences" of the fastaread block to the "Names" optional input port of seqlinkageBlock to label the leave nodes of the output phylogenetic tree.
connect(P,"fastaread","seqlinkage",["sequences","Names"]);
Optionally, you can also pass in a list of node names as the value of the optional input port "Names" by setting the property seqlinkageBlock.Inputs.Names.Value.
Before you run the pipeline, ensure that the folder of the class definition file is on the MATLAB search path. The reason is because each pipeline block runs in its own folder, and the external class definition files or function files will not be detected by the pipeline unless they are on the path.
% Update the following addpath call to specify the path to your class definition file. For instance, if Seqlinkage.m is located in C:\Examples\SubclassExample\, use the following: addpath("C:\Examples\SubclassExample\");
Run the pipeline.
run(P);
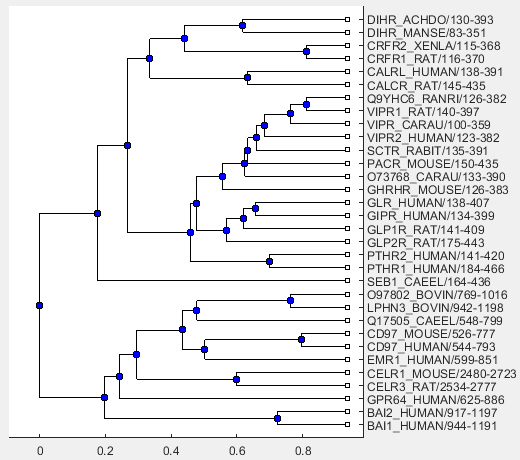
Get the result from seqlinkageBlock, which contains a phylogenetic tree object.
linkageResult = fetchResults(P,seqlinkageBlock)
linkageResult = struct with fields:
Phytree: [1×1 phytree]
Use the following command to visualize the phylogenetic tree.
view(linkageResult.Phytree)
Version History
Introduced in R2023a