connect
Syntax
Description
connect(
connects an output port of the pipeline,sourceBlock,targetBlock,newPortMap)sourceBlock to an input port of the
targetBlock. newPortMap specifies the output
and input ports.
mappedPorts = connect(pipeline,sourceBlock,targetBlock,newPortMap)mappedPorts between the
source block and target block after connecting two blocks as specified by
newPortMap.
Examples
Input Arguments
Bioinformatics pipeline, specified as a bioinfo.pipeline.Pipeline object.
Block in the pipeline to connect from, specified as a bioinfo.pipeline.Block object or character vector or string scalar that
represents the block name.
Block in the pipeline to connect to, specified as a bioinfo.pipeline.Block object or character vector or string scalar that
represents the block name.
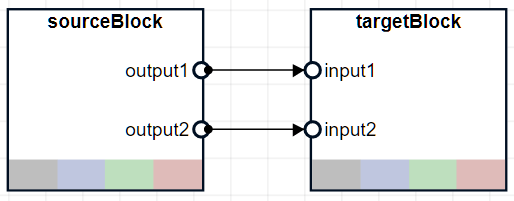
Output port name of the source block and an input port name of the target block to connect, specified an N-by-2 string array, where N is the number of connections between two blocks.
For example, if you are connecting the source block that has two output ports,
"output1" and "output2", to the target block
that has two input ports, "input1" and "input2",
specify newPortMap as a 2-by-2 string array:
["output1","input1";"output2","input2"]. The next figure is a
graphical representation of such connections.
Tip
To get the names of the input or output ports of a block, check the
Inputs and Outputs properties of the
block. Each property is returned as a structure, where each field name represents the
corresponding port name that you can use to connect the blocks.
Data Types: string
Output Arguments
Version History
Introduced in R2023a