processTable
Description
Examples
Input Arguments
Bioinformatics pipeline, specified as a bioinfo.pipeline.Pipeline object.
Pipeline blocks, specified as a bioinfo.pipeline.Block object, vector of block objects, character vector,
string scalar, string vector, or cell array of character vectors representing block
names.
Flag to contain one row per each independent block run or group all runs of the same
block into a single row, specified as a numeric or logical 1 (true)
or 0 (false).
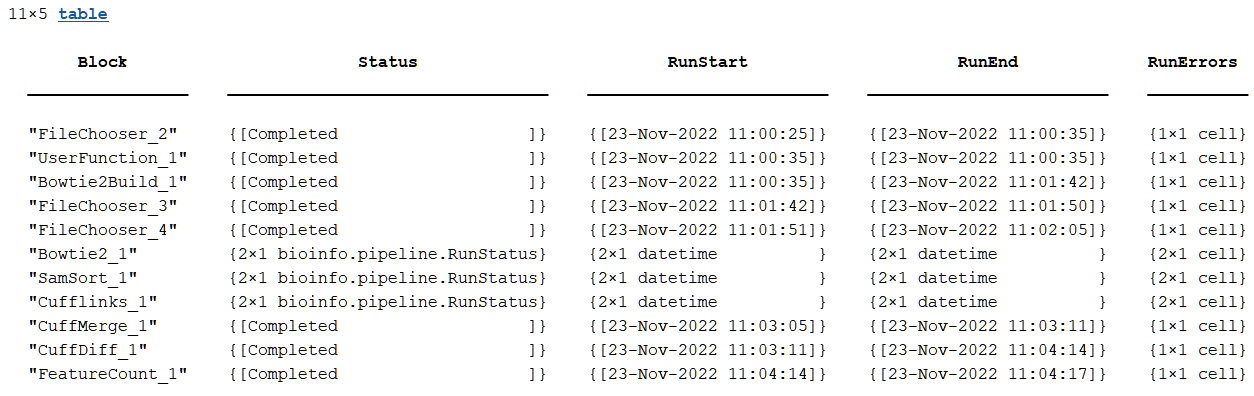
Each row in the table represents a block, and there are corresponding Status, RunStart, RunEnd, and RunErrors columns for each block.
By default, the function groups (or collapses) all runs of the same block into a
single row. If a block is run n independent times (per its
SplitDimension property of the input port), then the table
variable value for each column is an n-by-1 cell array. For instance,
the next figure is an example of a (default) collapsed process table (Expanded
= false). Note that for the Bowtie2_1,
SamSort_1 and Cufflinks_1 blocks, the table
variable values are 2-by-1 cell arrays because each of these blocks were run 2 times
independently.
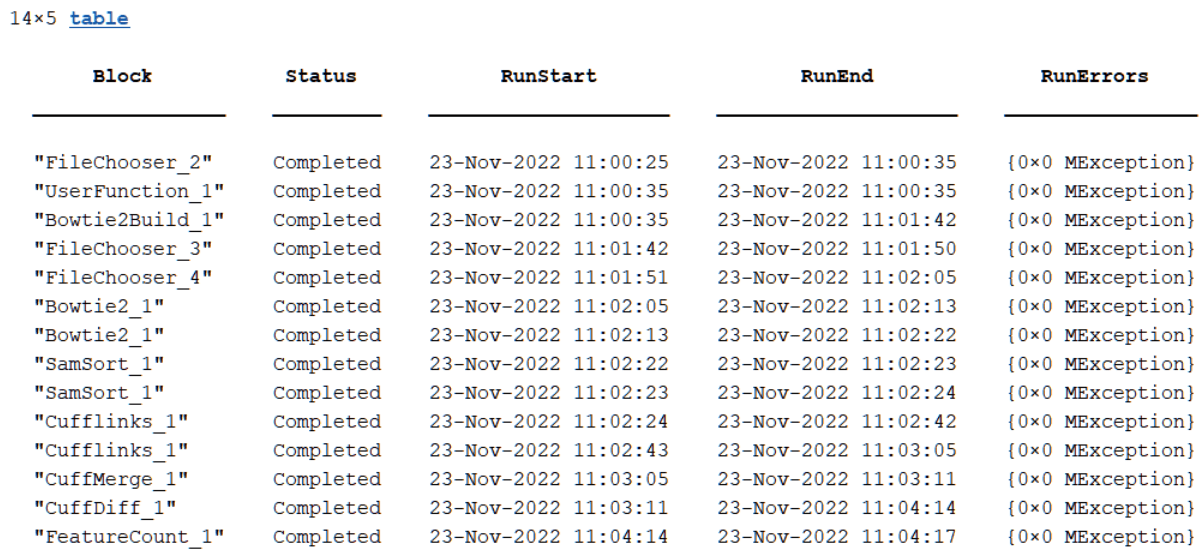
The next figure is the same table with the expanded cell array (Expanded =
true).
Output Arguments
Version History
Introduced in R2023a