SRASAMDumpOptions
Description
An SRASAMDumpOptions object contains options for the srasamdump
function, which you use to download the files from SRA (Sequence Read Archive) [1].
Creation
Description
sraOpt = SRASAMDumpOptionsSRASAMDumpOptions object with default property values.
SRASAMDumpOptions requires the SRA Toolkit for Bioinformatics Toolbox™. If this support package is not installed, then the function provides a download
link. For details, see Bioinformatics Toolbox Software Support Packages.
sraOpt = SRASAMDumpOptions(Name=Value)SRASAMDumpOptions object, specify BZip2=true to set
the value of the BZip2 property to true, so that
the output files are compressed using bzip2.
sraOpt = SRASAMDumpOptions(S)S.
Input Arguments
Properties
Object Functions
getCommand | Translate object properties to original options syntax |
getOptionsTable | Return table with all properties and equivalent options in original syntax |
Examples
Download some paired-end sequencing data in a FASTQ format using an accession run number SRR11846824 that has two reads per spot and has no unaligned reads. Downloading the data may take a few minutes.
tbl = srafasterqdump("SRR11846824")tbl=1×2 table
"SRR11846824_1.fastq" "SRR11846824_2.fastq"
By default, the function uses the SplitType="SplitThree" option and downloads only biological reads. Specifically, the function splits spots into reads. For spots having two reads, the function produces *_1.fastq and *_2.fastq, represented by the Reads_1 and Reads_2 columns. If there are any unaligned reads, the function saves unaligned reads in a *.fastq file, which would be represented by the Reads column. Because there are no unaligned reads within this accession, the function did not produce a *.fastq file, and the output table has no Reads column. For details, see SplitType.
You can also specify other download options using SRAFasterqDumpOptions. For instance, use FastaOutput=true to get the FASTA-formatted file.
sraopt = SRAFasterqDumpOptions;
sraopt.FastaOutput = true;
tbl2 = srafasterqdump("SRR11846824",sraopt);Alternatively, you can specify the options as name-value arguments instead of using the options object.
tbl2 = srafasterqdump("SRR11846824",FastaOutput=true);
You can also download the data in a SAM format using srasamdump.
samFile = srasamdump("SRR11846824")samFile = "SRR11846824.sam"
Specify the download options using an SRASAMDumpOptions object. For instance, specify the output file name and compress the output file using bzip2.
samdumpopt = SRASAMDumpOptions;
samdumpopt.OutputFileName = "SRR11846824.sam.bz2";
samdumpopt.BZip2 = truesamdumpopt =
SRASAMDumpOptions with properties:
Default properties:
ExtraCommand: ""
FastaOutput: 0
FastqOutput: 0
GZip: 0
HideIdentical: 0
IncludeAll: 0
MinMapQuality: 0
OutputPrimary: 0
OutputUnaligned: 0
Version: "3.0.6"
Modified properties:
OutputFileName: "SRR11846824.sam.bz2"
BZip2: 1
bzFile = srasamdump("SRR11846824",samdumpopt)bzFile = "SRR11846824.sam.bz2"
After downloading the SAM file, you can use it for downstream analyses. For instance, you can use bowtie2 to map the reads to the reference sequence.
First, download the C. elegans reference sequence.
celegans_refseq = fastaread("https://s3.amazonaws.com/igv.broadinstitute.org/genomes/seq/ce11/ce11.fa");Save Chromosome 3 reference data in a FASTA file.
celegans_chr3 = celegans_refseq(3).Sequence; warnState = warning; warning('off','Bioinfo:fastawrite:AppendToFile'); fastawrite("celegans_chr3.fa",celegans_chr3); warning(warnState);
Build a set of index files using bowtie2build. The status value of 0 means that the build was successful.
status = bowtie2build("celegans_chr3.fa","celegans_chr3_index");
Align read data to the reference. This may take a few minutes.
bowtie2("celegans_chr3_index","SRR11846824_1.fastq","SRR11846824_2.fastq","SRR11846824_mapped.sam");
Create a quality control plot for the SAM file. Note that, for this particular experiment, most of the reads happen to have the same quality score of 30.
seqqcplot("SRR11846824_mapped.sam");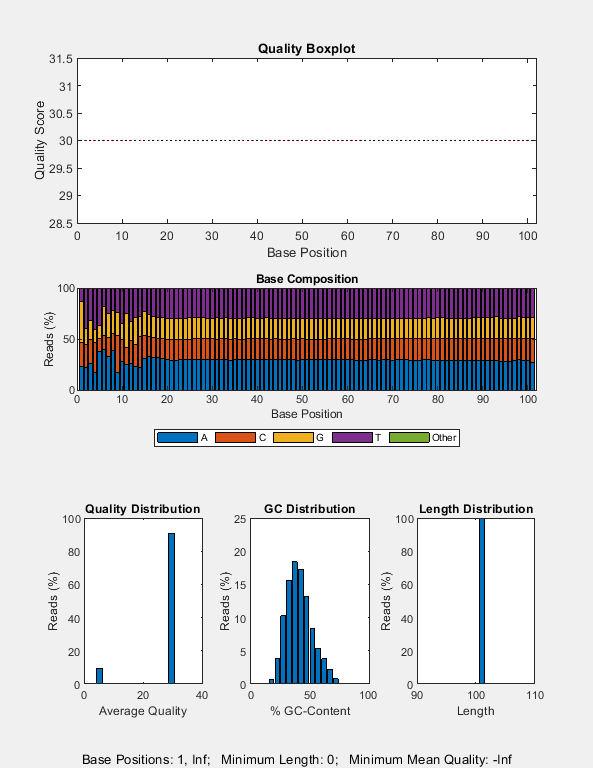
Convert the SAM file to a BAM file. Suppress two informational warnings that are issued while creating a BioMap object.
w = warning; warning("off","bioinfo:BioMap:BioMap:UnsortedReadsInSAMFile"); warning("off","bioinfo:saminfo:InvalidTagField"); bmObj = BioMap("SRR11846824_mapped.sam"); write(bmObj,"SRR11846824_mapped.bam",Format="BAM"); warning(w);
Visualize the alignment data in the Genomics Viewer app. The corresponding cytoband file is provided with the toolbox.
gv = genomicsViewer(ReferenceFile="celegans_chr3.fa",CytoBand="celegans_cytoBandIdeo.txt.gz"); addTracks(gv,"SRR11846824_mapped.bam");
Use the zoom slider to zoom in and see the features. Or you can enter the following in the search text box: Generated:3,711,861-3,711,940.
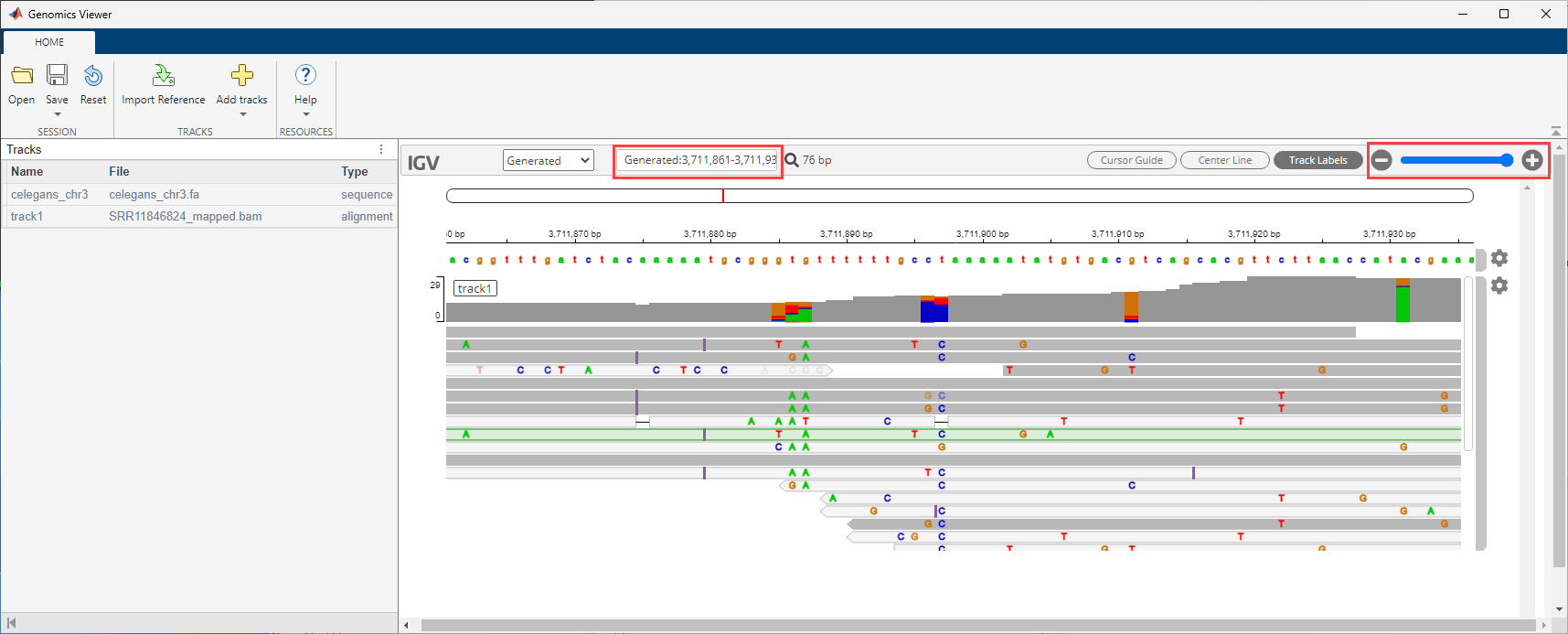
You may delete the downloaded files, such as the reference sequence file.
delete celegans_chr3.faClose the app.
close(gv);
References
[1] SRA Toolkit Development Team https://github.com/ncbi/sra-tools/wiki/01.-Downloading-SRA-Toolkit
Version History
Introduced in R2024a