predict
Simulate and evaluate fitted SimBiology model
Syntax
Description
[
returns simulation results ynew,parameterEstimates]
= predict(resultsObj)ynew from evaluating the fitted SimBiology® model. The estimated parameter values parameterEstimates
used to compute ynew are from the original fit by
sbiofitmixed.
[
returns simulation results ynew,parameterEstimates]
= predict(resultsObj,data,dosing)ynew from evaluating the fitted SimBiology model by using the specified data and
dosing information.
[
uses additional options specified by one or more name-value arguments.ynew,parameterEstimates]
= predict(___,Name,Value)
Tip
Use this method to get model responses at specific time points or to predict model responses using different covariate data and dosing information.
Examples
Estimate nonlinear mixed-effects parameters using clinical pharmacokinetic data collected from 59 infants. Evaluate the fitted model given new data or dosing information.
Load Data
This example uses data collected on 59 preterm infants given phenobarbital during the first 16 days after birth [1]. ds is a table containing the concentration-time profile data and covariate information for each infant (or group).
load pheno.mat ds
Convert to groupedData
Convert the data to the groupedData format for parameter estimation.
data = groupedData(ds);
Display the first few rows of data.
data(1:5,:)
ans =
5×6 groupedData
ID TIME DOSE WEIGHT APGAR CONC
__ ____ ____ ______ _____ ____
1 0 25 1.4 7 NaN
1 2 NaN 1.4 7 17.3
1 12.5 3.5 1.4 7 NaN
1 24.5 3.5 1.4 7 NaN
1 37 3.5 1.4 7 NaN
Visualize Data
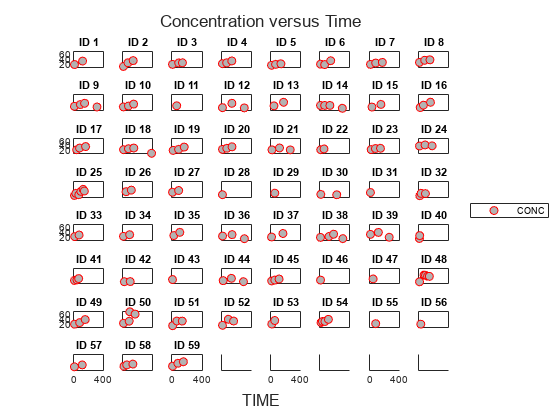
Display the data in a trellis plot.
t = sbiotrellis(data, 'ID', 'TIME', 'CONC', 'marker', 'o',... 'markerfacecolor', [.7 .7 .7], 'markeredgecolor', 'r', ... 'linestyle', 'none'); t.plottitle = 'Concentration versus Time';
Create a One-Compartment PK Model
Create a simple one-compartment PK model, with bolus dose administration and linear clearance elimination, to fit the data.
pkmd = PKModelDesign; addCompartment(pkmd,'Central','DosingType','Bolus',... 'EliminationType','linear-clearance',... 'HasResponseVariable',true,'HasLag',false); onecomp = pkmd.construct;
Map model species to response data.
responseMap = 'Drug_Central = CONC';Define Estimated Parameters
The parameters to estimate in this model are the volume of the central compartment (Central) and the clearance rate (Cl_Central). sbiofitmixed calculates fixed and random effects for each parameter. The underlying algorithm computes normally distributed random effects, which might violate constraints for biological parameters that are always positive, such as volume and clearance. Therefore, specify a transform for the estimated parameters so that the transformed parameters follow a normal distribution. The resulting model is
and
where , , and are the fixed effects, random effects, and estimated parameter values respectively, calculated for each infant (group) . Some arbitrary initial estimates for V (volume of central compartment) and Cl (clearance rate) are used here in the absence of better empirical data.
estimatedParams = estimatedInfo({'log(Central)','log(Cl_Central)'},'InitialValue',[1 1]);Define Dosing
All infants were given the drug, represented by the Drug_Central species, where the dosing schedule varies among infants. The amount of drug is listed in the data variable DOSE. You can automatically generate dose objects from the data and use them during fitting. In this example, Drug_Central is the target species that receives the dose.
sampleDose = sbiodose('sample','TargetName','Drug_Central'); doses = createDoses(data,'DOSE','',sampleDose);
Fit the Model
Use sbiofitmixed to fit the one-compartment model to the data.
nlmeResults = sbiofitmixed(onecomp,data,responseMap,estimatedParams,doses,'nlmefit');Visualize Results
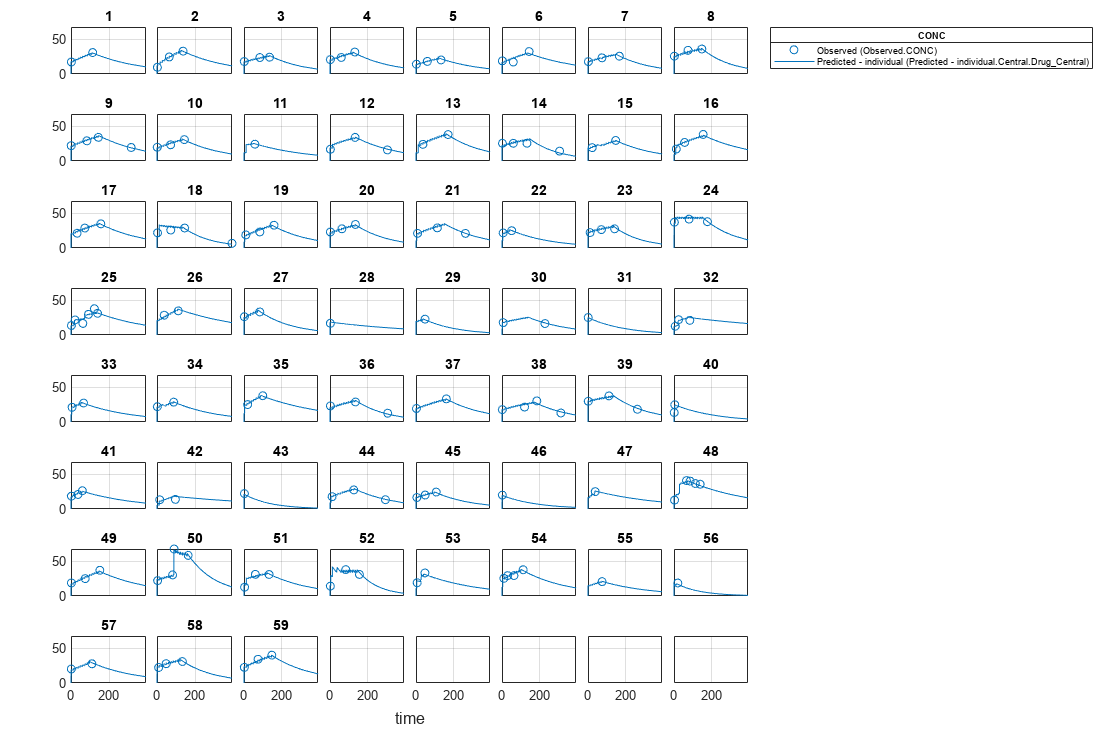
Visualize the fitted results using individual-specific parameter estimates.
plot(nlmeResults,'ParameterType','individual');
Use New Dosing Data to Simulate the Fitted Model
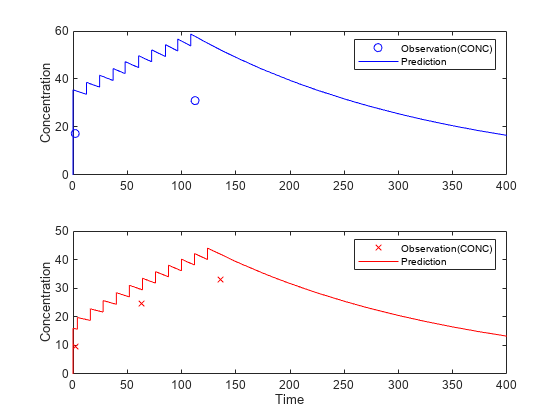
Suppose you want to predict how infants 1 and 2 would have responded under different dosing amounts. You can predict their responses as follows.
Create new dose objects with new dose amounts.
dose1 = doses(1); dose1.Amount = dose1.Amount*2; dose2 = doses(2); dose2.Amount = dose2.Amount*1.5;
Use the predict function to evaluate the fitted model using the new dosing data. If you want response predictions at particular times, provide the new output time vector. Use the 'ParameterType' option to specify individual or population parameters to use. By default, predict uses the population parameters when you specify output times.
timeVec = [0:25:400]; newResults = predict(nlmeResults,timeVec,[dose1;dose2],'ParameterType','population');
Visualize the predicted responses while overlapping the experimental data for infants 1 and 2.
figure; subplot(2,1,1) plot(data.TIME(data.ID == 1),data.CONC(data.ID == 1),'bo') hold on plot(newResults(1).Time,newResults(1).Data,'b') hold off ylabel('Concentration') legend('Observation(CONC)','Prediction') subplot(2,1,2) plot(data.TIME(data.ID == 2),data.CONC(data.ID == 2),'rx') hold on plot(newResults(2).Time,newResults(2).Data,'r') hold off legend('Observation(CONC)','Prediction') ylabel('Concentration') xlabel('Time')
Create a Covariate Model for the Covariate Dependencies
Suppose there is a correlation between volume and weight, and possibly volume and APGAR score. Consider the effect of weight by modeling two of these covariate dependencies: the volume of central (Central) and the clearance rate (Cl_Central) vary with weight. The model becomes
and
Use the CovariateModel object to define the covariate dependencies. For details, see Specify a Covariate Model.
covModel = CovariateModel;
covModel.Expression = ({'Central = exp(theta1 + theta2*WEIGHT + eta1)',...
'Cl_Central = exp(theta3 + theta4*WEIGHT + eta2)'});Use constructDefaultInitialEstimate to create an initialEstimates struct.
initialEstimates = covModel.constructDefaultFixedEffectValues;
Use the FixedEffectNames property to display the thetas (fixed effects) defined in the model.
covModel.FixedEffectNames
ans = 4×1 cell
{'theta1'}
{'theta3'}
{'theta2'}
{'theta4'}
Use the FixedEffectDescription property to show the descriptions of corresponding fixed effects (thetas) used in the covariate expression. For example, theta2 is the fixed effect for the weight covariate that correlates with the volume (Central), denoted as 'Central/WEIGHT'.
disp('Fixed Effects Description:');Fixed Effects Description:
disp(covModel.FixedEffectDescription);
{'Central' }
{'Cl_Central' }
{'Central/WEIGHT' }
{'Cl_Central/WEIGHT'}
Set the initial guesses for the fixed-effect parameter values for Central and Cl_Central using the values estimated from fitting the base model.
initialEstimates.theta1 = nlmeResults.FixedEffects.Estimate(1); initialEstimates.theta3 = nlmeResults.FixedEffects.Estimate(2); covModel.FixedEffectValues = initialEstimates;
Fit the Model
nlmeResults_cov = sbiofitmixed(onecomp,data,responseMap,covModel,doses,'nlmefit');Display Fitted Parameters and Covariances
disp('Estimated Fixed Effects:');Estimated Fixed Effects:
disp(nlmeResults_cov.FixedEffects);
Name Description Estimate StandardError
__________ _____________________ ________ _____________
{'theta1'} {'Central' } -0.45664 0.078933
{'theta3'} {'Cl_Central' } -5.9519 0.1177
{'theta2'} {'Central/WEIGHT' } 0.52948 0.047342
{'theta4'} {'Cl_Central/WEIGHT'} 0.61954 0.071386
disp('Estimated Covariance Matrix:');Estimated Covariance Matrix:
disp(nlmeResults_cov.RandomEffectCovarianceMatrix);
eta1 eta2
________ ________
eta1 0.046503 0
eta2 0 0.041609
Visualize Results
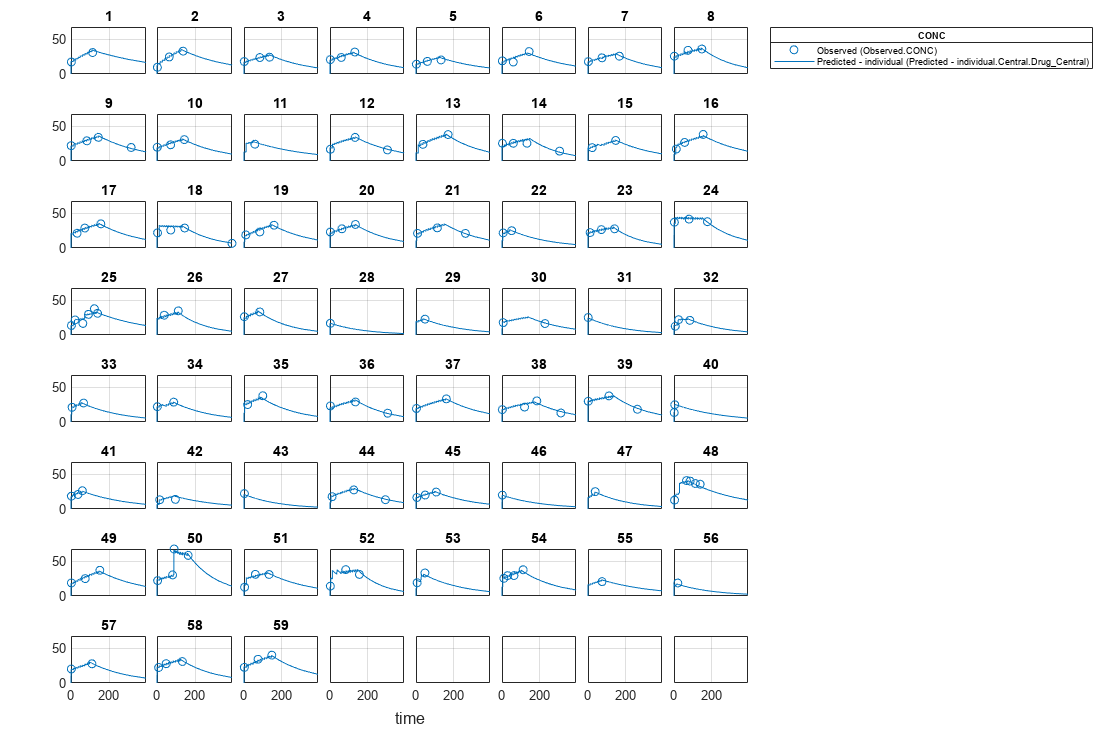
Visualize the fitted results using individual-specific parameter estimates.
plot(nlmeResults_cov,'ParameterType','individual');
Use New Covariate Data to Evaluate the Fitted Model
Suppose you want to explore the responses of infants 1 and 2 using different covariate data, namely WEIGHT. You can do this by specifying the new WEIGHT data. The ID variable of the data corresponds to individual infants.
newData = data(data.ID == 1 | data.ID == 2,:); newData.WEIGHT(newData.ID == 1) = 1.3; newData.WEIGHT(newData.ID == 2) = 1.4;
Simulate the responses of infants 1 and 2 using the new covariate data.
[newResults_cov, newEstimates] = predict(nlmeResults_cov,newData,[dose1;dose2]);
newEstimates contains the updated parameter estimates for each individual (infants 1 and 2) after the model is reevaluated using the new covariate data.
newEstimates
newEstimates=4×3 table
Group Name Estimate
_____ ______________ _________
1 {'Central' } 2.5596
1 {'Cl_Central'} 0.0065965
2 {'Central' } 1.7123
2 {'Cl_Central'} 0.0064806
Compare to the estimated values from the original fit using the old covariate data.
nlmeResults_cov.IndividualParameterEstimates( ... nlmeResults_cov.IndividualParameterEstimates.Group == '1' | ... nlmeResults_cov.IndividualParameterEstimates.Group == '2',:)
ans=4×3 table
Group Name Estimate
_____ ______________ _________
1 {'Central' } 2.6988
1 {'Cl_Central'} 0.0070181
2 {'Central' } 1.8054
2 {'Cl_Central'} 0.0068948
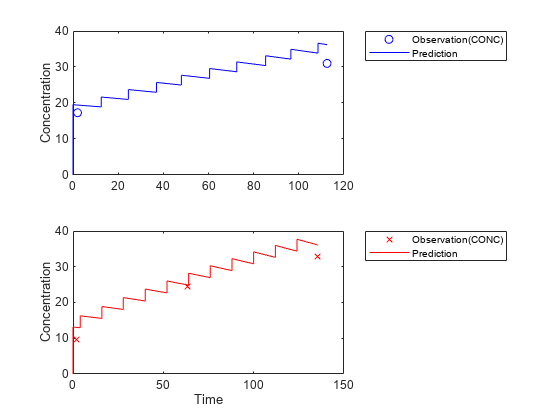
Visualize the new simulation results together with the experimental data for infant 1 and 2.
figure; subplot(2,1,1); plot(data.TIME(data.ID == 1),data.CONC(data.ID == 1),'bo') hold on plot(newResults_cov(1).Time,newResults_cov(1).Data,'b') hold off ylabel('Concentration') legend('Observation(CONC)','Prediction','Location','NorthEastOutside') subplot(2,1,2) plot(data.TIME(data.ID == 2),data.CONC(data.ID == 2),'rx') hold on plot(newResults_cov(2).Time,newResults_cov(2).Data,'r') hold off legend('Observation(CONC)','Prediction','Location','NorthEastOutside') ylabel('Concentration') xlabel('Time')
References
[1] Grasela, T. H. Jr., and S. M. Donn. "Neonatal population pharmacokinetics of phenobarbital derived from routine clinical data." Dev Pharmacol Ther 1985:8(6). 374-83.
Input Arguments
Name-Value Arguments
Output Arguments
Version History
Introduced in R2014a