msalign
Align peaks in signal to reference peaks
Syntax
Description
IntensitiesOut = msalign(X,Intensities,RefX)Intensities and
X, to reference peaks, provided by RefX.
IntensitiesOut = msalign(X,Intensities,RefX,Name,Value)msalign using one or more
Name=Value arguments. For example, you can have msalign
take more iterations than the default five by specifying
Iterations=10.
[
also returns IntensitiesOut,RefXOut] = msalign(X,Intensities,RefX,Name,Value)RefXout, a new vector of separation-unit values to use as
reference masses for aligning the peaks. RefXOut differs from
RefX only when the Group name-value argument is
true.
Examples
Load the sample_lo_res file, which is included with the toolbox.
load sample_lo_resSet the markers vector and the weights vector.
R = [3991.4 4598 7964 9160]; W = [60 100 60 100];
Display a color image of the mass spectra before alignment.
msheatmap(MZ_lo_res,Y_lo_res,'markers',R,'range',[3000 10000]) title('before alignment')
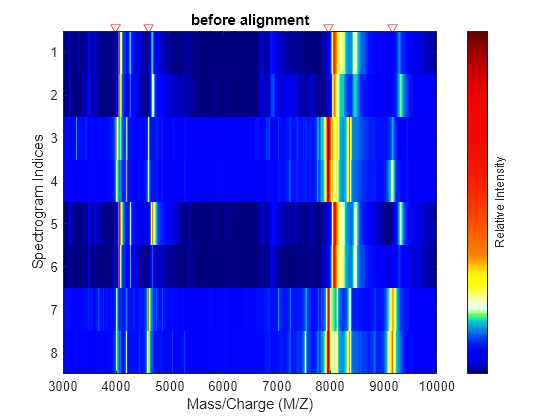
Align spectra with reference masses and display a color image of mass spectra after alignment.
YA = msalign(MZ_lo_res,Y_lo_res,R,'weights',W); msheatmap(MZ_lo_res,YA,'markers',R,'range',[3000 10000]) title('after alignment')
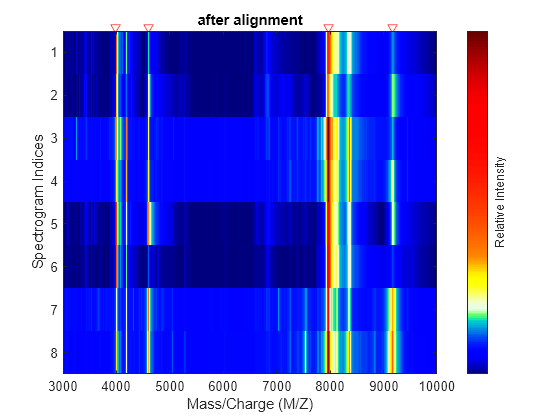
Now the spectrogram displays much better vertical alignment.
If you have only one reference peak in your data, do not use the msalign function. Instead, use the following procedure, which shifts, but does not scale, the X input vector.
Load the sample_lo_res data and view the first sample spectrum.
load sample_lo_res
MZ = MZ_lo_res;
Y = Y_lo_res(:,1);
msviewer(MZ,Y)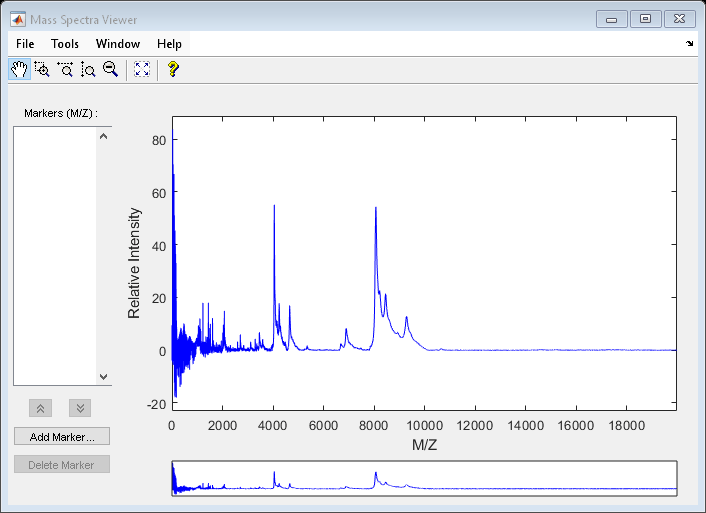
Use the tall peak around 4000 m/z as the reference peak. To determine the reference peak's m/z value, click ![]() , and then click-drag to zoom in on the peak. Right-click in the center of the peak, and then click Add Marker to label the peak with its m/z value.
, and then click-drag to zoom in on the peak. Right-click in the center of the peak, and then click Add Marker to label the peak with its m/z value.
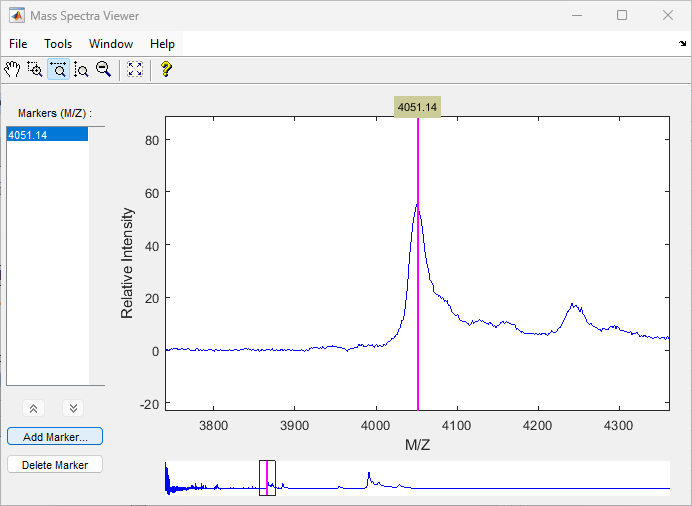
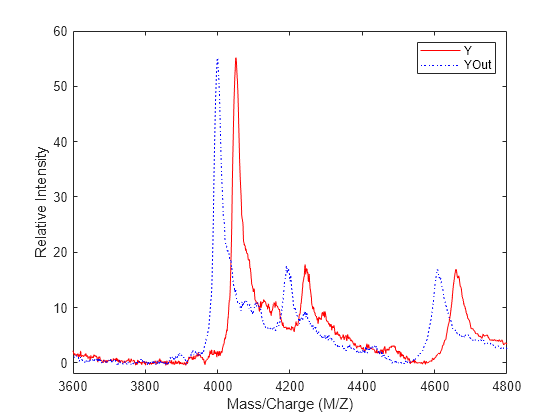
Shift a spectrum by the difference between RP, the known reference mass of 4000 m/z, and SP, the experimental mass of 4051.14 m/z.
RP = 4000; SP = 4051.14; YOut = interp1(MZ, Y, MZ-(RP-SP));
Plot the original spectrum in red and the shifted spectrum in blue and zoom in on the reference peak.
plot(MZ,Y,'r',MZ,YOut,'b:') xlabel('Mass/Charge (M/Z)') ylabel('Relative Intensity') legend('Y','YOut') axis([3600 4800 -2 60])
Input Arguments
Name-Value Arguments
Output Arguments
Algorithms
First, msalign creates a synthetic signal from the reference peaks
using Gaussian pulses centered at the separation-unit values specified by
RefX. Then, msalign shifts and scales the
separation-unit scale to find the maximum alignment between the input signals and the synthetic
signal. (msalign uses an iterative multiresolution grid search until it
finds the best scale and shift factors for each signal.) Once msalign
determines the new separation-unit scale, msalign creates the corrected
signals by resampling their intensities at the original separation-unit values, creating
IntensitiesOut, a vector or matrix of corrected intensity values. The
resampling method preserves the shape of the peaks.
References
[1] Monchamp, P., Andrade-Cetto, L., Zhang, J.Y., and Henson, R. (2007) Signal Processing Methods for Mass Spectrometry. In Systems Bioinformatics: An Engineering Case-Based Approach, G. Alterovitz and M.F. Ramoni, eds. (Artech House Publishers).
Version History
Introduced before R2006a
See Also
mspalign | msbackadj | msdotplot | msheatmap | mslowess | msnorm | mspeaks | msresample | msppresample | mssgolay | msviewer
Topics
- Mass Spectrometry and Bioanalytics
- Preprocessing Raw Mass Spectrometry Data
- Visualizing and Preprocessing Hyphenated Mass Spectrometry Data Sets for Metabolite and Protein/Peptide Profiling
- Differential Analysis of Complex Protein and Metabolite Mixtures Using Liquid Chromatography/Mass Spectrometry (LC/MS)